A sourmash tutorial¶
sourmash is our lab’s implementation of an ultra-fast lightweight approach to nucleotide-level search and comparison, called MinHash.
You can read some background about MinHash sketches in this paper: Mash: fast genome and metagenome distance estimation using MinHash. Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM. Genome Biol. 2016 Jun 20;17(1):132. doi: 10.1186/s13059-016-0997-x.
Installing sourmash¶
To install sourmash, run:
sudo apt-get install -y python3.5-dev python3.5-venv
this installs Python 3.5.
Now, create a local software install and populate it with Jupyter and other dependencies:
python3.5 -m venv ~/py3
. ~/py3/bin/activate
pip install -U pip
pip install -U Cython
pip install -U jupyter jupyter_client ipython pandas matplotlib scipy scikit-learn
pip install https://github.com/dib-lab/khmer/archive/master.zip
pip install -U https://github.com/dib-lab/sourmash/archive/2017-ucsc-metagenome.zip
(Note, we are installing from a development branch; many of the features below are not part of an official sourmash release yet. They should be included in sourmash 2.0.)
Fingerprint reads¶
Compute a scaled MinHash fingerprint from our reads:
mkdir ~/sourmash
cd ~/sourmash
sourmash compute --scaled 10000 ~/data/ecoli_ref*pe*.fq.gz -o ecoli-reads.sig -k 31
Compare reads to assemblies¶
Use case: how much of the read content is contained in the assembly?
Fingerprint the assembly:
sourmash compute --scaled 10000 -k 31 ~/assembly/ecoli/final.contigs.fa -o ecoli-genome.sig
and now evaluate containment, that is, what fraction of the read content is contained in the assembly:
sourmash search -k 31 ecoli-reads.sig ecoli-genome.sig --containment
and you should see:
# running sourmash subcommand: search
loaded query: /home/ubuntu/data/ecoli_ref-5m... (k=31, DNA)
loading db of signatures from 1 files
loaded 1 signatures total.
1 matches; showing 3:
/home/ubuntu/assembly/ecoli/final.contigs.fa 0.466 ecoli-genome.sig
Try the reverse - why is it bigger?
sourmash search -k 31 ecoli-genome.sig ecoli-reads.sig --containment
Make and search a database quickly.¶
Suppose that we have a collection of signatures (made with sourmash compute as above) and we want to search it with our newly assembled
genome (or the reads, even!). How would we do that?
Let’s grab a sample collection of 50 E. coli genomes and unpack it –
mkdir ecoli_many_sigs
cd ecoli_many_sigs
curl -O -L https://github.com/ngs-docs/2017-lapaz-assembly/raw/master/_static/eschericia-sigs.tar.gz
tar xzf eschericia-sigs.tar.gz
cd ../
This will produce 50 files named ecoli-N.sig in the ecoli_many_sigs –
ls ecoli_many_sigs
Let’s turn this into an easily-searchable database with sourmash sbt_index –
sourmash sbt_index -k 31 ecolidb ecoli_many_sigs/*.sig
and now we can search!
sourmash sbt_search ecolidb.sbt.json ecoli-genome.sig | head
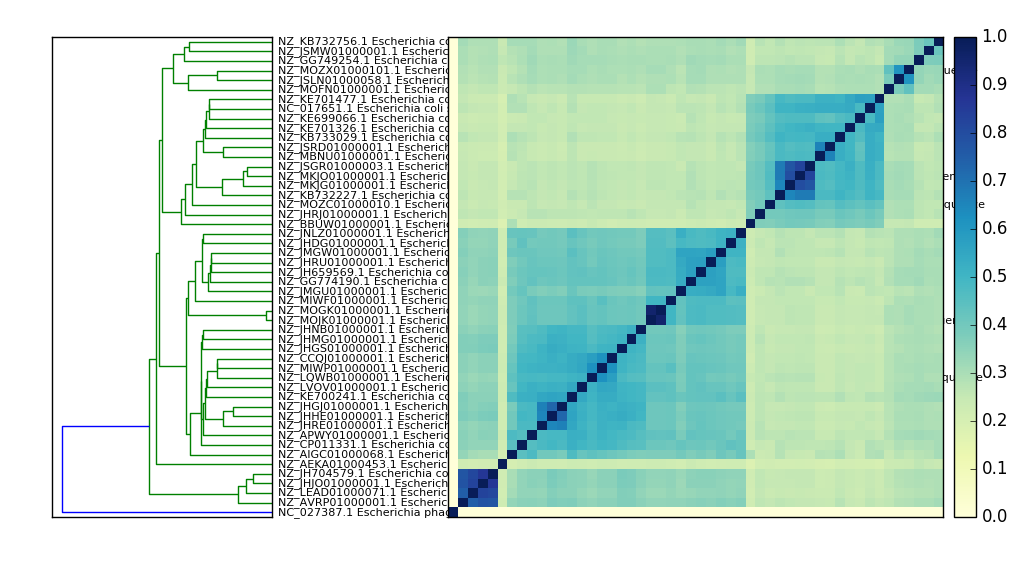
Compare many signatures and build a tree.¶
Adjust plotting (this is a bug in sourmash :) –
echo 'backend : Agg' > matplotlibrc
Compare all the things:
sourmash compare ecoli_many_sigs/* -o ecoli_cmp
and then plot:
sourmash plot --pdf --labels ecoli_cmp
which will produce a file ecoli_cmp.matrix.pdf and ecoli_cmp.dendro.pdf
which you can then download via your file browser and view on your local
computer.
Here’s a PNG version:
What’s in my metagenome?¶
Download and unpack a newer version of the k=31 RefSeq index described in CTB’s blog post:
curl -O https://s3-us-west-1.amazonaws.com/spacegraphcats.ucdavis.edu/microbe-genbank-sbt-k31-2017.05.09.tar.gz
tar xzf microbe-genbank-sbt-k31-2017.05.09.tar.gz
This produces a file genbank-k31.sbt.json and a whole bunch of hidden
files in the directory .sbt.genbank-k31. This is an index of about 100,000
microbial genomes from RefSeq.
Next, run the ‘gather’ command to see what’s in there –
sourmash sbt_gather -k 31 genbank-k31.sbt.json ecoli-genome.sig
and you should get:
# running sourmash subcommand: sbt_gather
loaded query: /home/ubuntu/assembly/ecoli/fi... (k=31, DNA)
overlap p_query p_genome
------- ------- --------
4.9 Mbp 99.8% 99.8% APIN01000001.1 Escherichia coli str. K-1
found less than 10.0 kbp in common. => exiting
found 1 matches total;
the recovered matches hit 100.0% of the query
You can use this on metagenomes (assembled and unassembled) as well; you’ve just got to make the signature files.
To see this in action, here is gather running on a signature generated from some sequences that assemble (but don’t align to known genomes) from the Shakya et al. 2013 mock metagenome paper.
curl -O -L https://github.com/ngs-docs/2017-lapaz-assembly/raw/master/_images/shakya-unaligned-contigs.sig
sourmash sbt_gather -k 31 genbank-k31.sbt.json shakya-unaligned-contigs.sig
This should yield:
# running sourmash subcommand: sbt_gather
loaded query: mqc500.QC.AMBIGUOUS.99.unalign... (k=31, DNA)
overlap p_query p_genome
------- ------- --------
1.4 Mbp 11.0% 58.0% JANA01000001.1 Fusobacterium sp. OBRC1 c
1.0 Mbp 7.7% 25.9% CP001957.1 Haloferax volcanii DS2 plasmi
0.9 Mbp 7.5% 11.8% BA000019.2 Nostoc sp. PCC 7120 DNA, comp
0.7 Mbp 5.9% 23.0% FOVK01000036.1 Proteiniclasticum ruminis
0.7 Mbp 5.3% 17.6% AE017285.1 Desulfovibrio vulgaris subsp.
0.6 Mbp 4.9% 11.1% CP001252.1 Shewanella baltica OS223, com
0.6 Mbp 4.8% 27.3% AP008226.1 Thermus thermophilus HB8 geno
0.6 Mbp 4.4% 11.2% CP000031.2 Ruegeria pomeroyi DSS-3, comp
480.0 kbp 3.8% 7.6% CP000875.1 Herpetosiphon aurantiacus DSM
410.0 kbp 3.3% 10.5% CH959317.1 Sulfitobacter sp. NAS-14.1 sc
1.4 Mbp 10.9% 11.8% LN831027.1 Fusobacterium nucleatum subsp
0.5 Mbp 4.1% 5.3% CP000753.1 Shewanella baltica OS185, com
420.0 kbp 3.3% 7.7% FNDZ01000023.1 Proteiniclasticum ruminis
150.0 kbp 1.2% 4.5% CP015081.1 Deinococcus radiodurans R1 ch
150.0 kbp 1.2% 8.2% CP000969.1 Thermotoga sp. RQ2, complete
290.0 kbp 2.3% 4.1% CH959311.1 Sulfitobacter sp. EE-36 scf_1
1.2 Mbp 9.4% 5.0% CP013328.1 Fusobacterium nucleatum subsp
110.0 kbp 0.9% 3.5% FREL01000833.1 Enterococcus faecalis iso
0.6 Mbp 5.0% 2.8% CP000527.1 Desulfovibrio vulgaris DP4, c
340.0 kbp 2.7% 3.3% KQ235732.1 Fusobacterium nucleatum subsp
70.0 kbp 0.6% 1.2% CP000850.1 Salinispora arenicola CNS-205
60.0 kbp 0.5% 0.7% CP000270.1 Burkholderia xenovorans LB400
50.0 kbp 0.4% 2.6% CP001080.1 Sulfurihydrogenibium sp. YO3A
50.0 kbp 0.4% 3.2% L77117.1 Methanocaldococcus jannaschii D
found less than 40.0 kbp in common. => exiting
found 24 matches total;
the recovered matches hit 73.4% of the query